Modelling tabular data with Google’s TabNet
Released in 2019, Google Research’s TabNet is claimed in a preprint manuscript to outperform existing methods on tabular data. How does it work and how can one try it?
Tabular data probably make up the majority of business data today. Think of things like retail transactions, click stream data, temperature and pressure sensors in factories, KYC information… the variety is endless.
In another post, I introduced CatBoost, one of my favorite methods for building prediction models on tabular data, and its neural network counterpart, NODE. But around the same time as the NODE manuscript came out, Google Research released a manuscript taking a totally different approach to tabular data modelling with neural networks. Whereas NODE mimics decision tree ensembles, Google’s proposed TabNet tries to build a new kind of architecture suitable for tabular data.
The paper describing the method is called TabNet: Attentive Interpretable Tabular Learning, which nicely summarizes what the authors are trying to do. The “Net” part tells us that it is a type of neural network, the “Attentive” part implies it is using an attention mechanism, it aims to be interpretable, and it is used for machine learning on tabular data.
How does it work?
TabNet uses a kind of soft feature selection to focus on just the features that are important for the example at hand. This is accomplished through a sequential multi-step decision mechanism. That is, the input information is processed top-down in several steps. As the manuscript puts it, “The idea of top-down attention in sequential form is inspired from its applications in processing visual and language data such as for visual question answering (Hudson & Manning, 2018) or in reinforcement learning (Mott et al., 2019) while searching for a small subset of relevant information in high dimensional input.”
The building blocks for performing this sequential attention are called transformer blocks even though they are a bit different from the transformers used in popular NLP models such as BERT. The soft feature selection is accomplished by using the sparsemax function.
The first figure from the paper, reproduced below, sketches how information is aggregated to form a prediction.
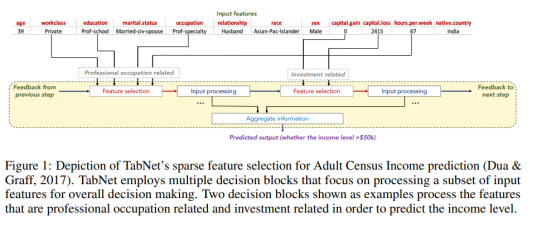
One nice property of TabNet is that it does not require feature preprocessing (in contrast to e.g. NODE). Another one is that it has interpretability built in “for free” in that the most relevant features are selected for each example. This means that you don’t have to apply an external explanation module such as shap or LIME.
It is not so easy to wrap one’s head around what is happening inside this architecture when reading the paper, but luckily there is published code which clarifies things a bit and shows that it is not as complicated as you might think.
How can I use it?
The original code and modifications
As already mentioned, the code is available, and the authors show how to use it together with the forest covertype dataset. To facilitate this, they have provided three dataset-specific files: one file that downloads and prepares the data (download_prepare_covertype.py), another one that defines the appropriate Tensorflow Feature Columns and a CSV reader input function (data_helper_covertype.py), and the file that contains the training loop (experiment_covertype.py).
The repo README states:
To modify the experiment to other tabular datasets:
– Substitute the train.csv, val.csv, and test.csv files under “data/” directory,
– Modify the data_helper function with the numerical and categorical features of the new dataset,
– Reoptimize the TabNet hyperparameters for the new dataset.
After having gone through this process a couple of times with other datasets, I decided to write my own wrapper code to streamline the process. This code, which I must stress is a totally unofficial fork, is on GitHub.
In terms of the README points above:
- Rather than making new train.csv, val.csv and test.csv files for each dataset, I preferred to read the entire dataset and do the splitting in-memory (as long as it is feasible, of course), so I wrote a new input function for Pandas in my code.
- It can take a bit of work to modify the data_helper.py file, at least initially when you aren’t quite sure what it does and how the feature columns should be defined (this was certainly the case with me). There are also many parameters which need to be changed but which are in the main training loop file rather than the data helper file. In view of this, I also tried to generalize and streamline this process in my code.
- I added some quick-and-dirty code for doing hyperparameter optimization, but so far only for classification.
- It is also worth mentioning that the example code from the authors only shows how to do classification, not regression, so that extra code also has to be written by the user. I have added regression functionality with a simple mean squared error loss.
Using the command-line interface
Execute a command like:
python train_tabnet.py \ --csv-path data/adult.csv \ --target-name "<=50K" \ --categorical-features workclass,education,marital.status,\ occupation,relationship,race,sex,native.country\ --feature_dim 16 \ --output_dim 16 \ --batch-size 4096 \ --virtual-batch-size 128 \ --batch-momentum 0.98 \ --gamma 1.5 \ --n_steps 5 \ --decay-every 2500 \ --lambda-sparsity 0.0001 \ --max-steps 7700
The mandatory parameters are — -csv-path(pointing to the location of the CSV file),--target-name(the name of the column with the prediction target) and--categorical-featues (a comma-separated list of the features that should be treated as categorical). The rest of the input parameters are hyperparameters that need to be optimized for each specific problem. The values shown above, though, are taken directly from the TabNet manuscript, so they have already been optimized for the Adult Census dataset by the authors.
By default, the training process will write information to the tflog subfolder of the location where you execute the script. You can point tensorboard at this folder to look at training and validation stats:
tensorboard --logdir tflog
and point your web browser to localhost:6006.
If you don’t have a GPU…
… you could try this Colaboratory notebook. Note that if you want to look at the Tensorboard logs, your best bet is probably to create a Google Storage bucket and have the script write the logs there. This is accomplished by using the tb-log-locationparameter. E.g. if your bucket’s name were camembert-skyscrape, you could add--tb-log-location gs://camembert-skyscraperto the invocation of the script. (Note, though, that you have to set the permissions for the storage bucket correctly. This can be a bit of a hassle.)
Then you can point tensorboard, from your own local computer, to that bucket:
tensorboard --logdir gs://camembert-skyscraper
Hyperparameter optimization
There is also a quick-and-dirty script for doing hyperparameter optimization in the repo (opt_tabnet.py). Again, an example is shown in the Colaboratory notebook. The script only works for classification so far, and it is worth noting that some training parameters are still hard-coded although they shouldn’t really be (for example, the patience parameter for early stopping [how many steps do you continue while the best validation accuracy does not improve].)
The parameters that are varied in the optimization script are N_steps, feature_dim, batch-momentum, gamma, lambda-sparsity. (output_dim is set to be equal to feature_dim, as suggested in the optimization tips just below.)
The paper has the following tips on hyperparameter optimization:
Most datasets yield the best results for N_steps ∈ [3, 10]. Typically, larger datasets and more complex tasks require a larger N_steps. A very high value of N_steps may suffer from overfitting and yield poor generalization.
Adjustment of the values of Nd [feature_dim] and Na [output_dim] is the most efficient way of obtaining a trade-off between performance and complexity. Nd = Na is a reasonable choice for most datasets. A very high value of Nd and Na may suffer from overfitting and yield poor generalization.
An optimal choice of γ can have a major role on the overall performance. Typically a larger N_steps value favors for a larger γ.
A large batch size is beneficial for performance — if the memory constraints permit, as large as 1–10 % of the total training dataset size is suggested. The virtual batch size is typically much smaller than the batch size.
Initially large learning rate is important, which should be gradually decayed until convergence.
Results
I’ve tried TabNet via this command line interface for several datasets, including the Adult Census dataset that I used in the post about NODE and CatBoost for reasons that can be found in that post. Conveniently, this dataset had also been used in the TabNet manuscript, and the authors present the best parameter settings they found there. With repeated runs using those setting, I noticed that the best validation error (and test error) tends to be at around 86%, similar to CatBoost without hyperparameter tuning. The authors report a test set performance of 85.7% in the manuscript. When I did hyperparameter optimization with hyperopt, I unsurprisingly reached a similar performance around 86%, albeit with a different parameter setting.
For other datasets such as the Poker Hand dataset, TabNet is claimed to beat other methods by a considerable margin. I have not yet devoted much time to that, but everyone is of course invited to try TabNet with hyperparameter optimization on various datasets for themselves!
Conclusions
TabNet is an interesting architecture that seems promising for tabular data analysis. It operates directly on raw data and uses a sequential attention mechanism to perform explicit feature selection for each example. This property also gives it a sort of built-in interpretability.
I have tried to make TabNet slightly easier to work with by writing some wrapper code around it. The next step is to compare it to other methods across a wide range of datasets.
Please try it on your own datasets and/or send pull requests and help me improve the interface if you are interested!